{kind=link}
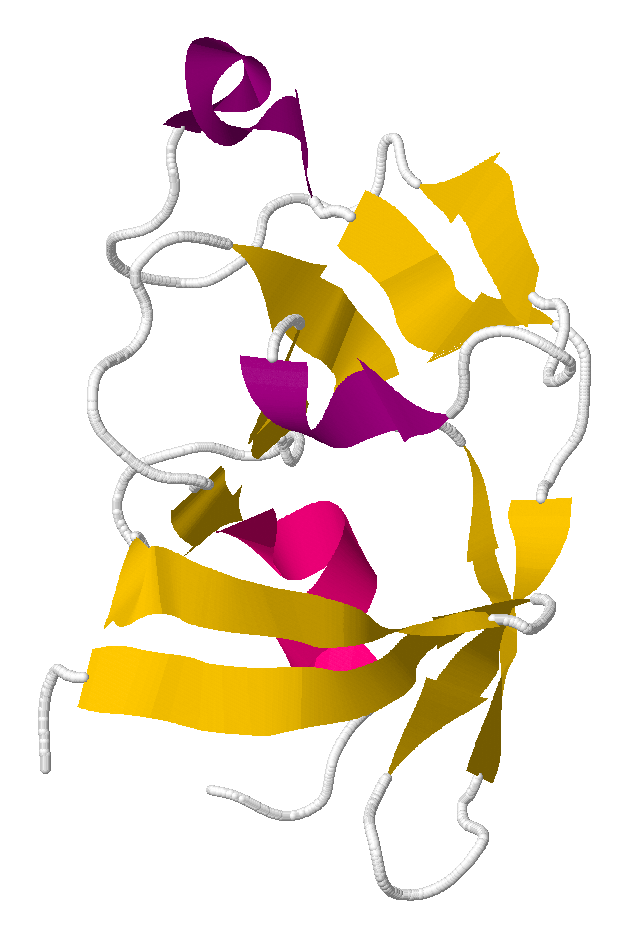
The solution to puzzle 390, published as PDB id 3SQF
Puzzle 390 involved a protein from Mason-Pfizer monkey virus. The successful results of this puzzle were a scientific breakthrough. Scientists had been unable to determine the correct structure of this protein for years. Foldit players figured it out in three weeks.
The Foldit page for Puzzle 390: Unsolved monkey virus protein has full results.
At the same time as puzzle 390, there were two beginner puzzles with the same protein, for players with less than:
The puzzle page described the original challenge:
The Folded monomer of protease from Mason-Pfizer monkey virus is currently unsolved by protein crystallographers. It was solved in 2003 by NMR, but there is a great deal of variation in those models and they don't fit perfectly with the X-ray crystallographic data. We are giving you these 10 NMR models to work on for 3 weeks, so that you have plenty of time to try out each model. We hope you can help us solve this structure!
It was a multi-start puzzle with 11 templates
The top score was 10,549 (evolver score by TheGUmmer).
Solutions[]
")
")
")
")
")
")
")
")
")
")
")
Discussion[]


{kind=link}
PDB id 1NSO, showing 10 solutions superimposed. The fan shape at the top means the solutions differ quite a bit.
beta_helix said "Can you come up with a model that crystallographers can use?
You can see the variation in the models solved by NMR in PDB id 1NSO
Our hope is that Foldit players can come up with a model that fits the more recent X-ray crystallographic data better than these NMR models from 2003. Then we could use that prediction for molecular replacement (http://en.wikipedia.org/wiki/Molecular_replacement) and solve this monkey virus protein using X-ray crystallography!
This would be an amazing scientific achievement, as we have been unable to use Rosetta to solve this particular structure using molecular replacement, but our lab has been able to do it with other proteins.
So we are giving you all 10 NMR models as starting structures (every time you reset the puzzle it will randomly select one of these 10 structures) and all 10 starts are also available in your Template Reserve in the Alignment Tool (as well as an extended chain conformation).
Even though the sequences for all 11 structures in the Alignment Tool are all identical, threading them will give you very different results. We hope that you can use partial threading to combine different regions from each NMR model.
Remember that by clicking on the 'Score' button on the top right of the Alignment Tool, you can see the name of each model ('model1', 'model2', etc., and 'extend' for the extended chain conformation) so that you can easily differentiate between the 10 different models. You can go back to the alignment score (which is useless for this puzzle since all the sequences are identical) by toggling the 'Name' button:" [1]
gramps gave partial threading instructions, then provided starting scores to identify which of the models is being used (along with LUA code snippet for future puzzles)
spvincent mentioned " Mike Tyka's beautiful protein blog. See http://beautifulproteins.blogspot.com/"
beta_helix said "In general, secondary structure composition can't be determined from unphased crystal data. There are some exceptions with all-helical proteins (http://www.nature.com/nmeth/journal/v6/n9/full/nmeth.1365.html) What we are hoping will work with Foldit is what was done previously using Rosetta: (http://www.nature.com/nature/journal/v450/n7167/abs/nature06249.html).
Most likely, the topology & secondary structure of the protein is pretty similar in the X-ray structure as in the NMR structure. Molecular replacement generally fails above 2A RMS so the target may not be very far from the NMR ensemble. That said, there may be some reorganization due to crystal contacts ... but sadly we really don't know."
Beta also said " you can download the pdf from the Baker Lab webpage:
http://depts.washington.edu/bakerpg/drupal/node/115
You'll find the Foldit nature paper there as well, if you didn't get a chance to read it:
http://depts.washington.edu/bakerpg/drupal/node/16
If you want to look at some X-ray structures that might be familiar to you, you can go to the CASP9 target page:
http://predictioncenter.org/casp9/targetlist.cgi
and click on the blue PDB code on the right for any of the targets that have been solved using X-ray crystallography (those are denoted by the 4th column on the CASP page saying "X-RAY")"